


‘Descoberta do ano’ da revista Science vai para inteligência artificial que prevê estrutura de proteínas
19 dezembro 2021 às 00h00
COMPARTILHAR
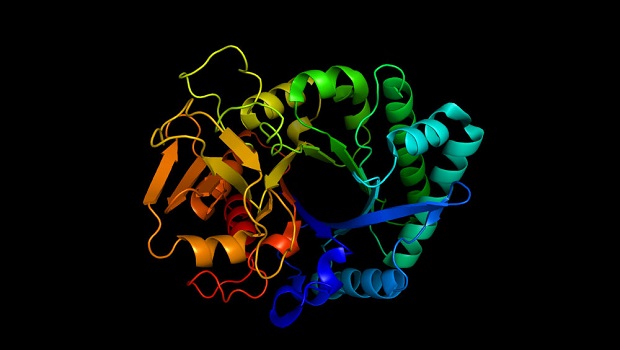
Softwares capazes de criar rapidamente modelos 3D de proteínas com longas cadeias de aminoácidos devem mudar o mundo da química e biologia molecular
Em 1972, ao aceitar seu Prêmio Nobel, o bioquímico americano Christian Anfinsen discursou: “Um dia será possível prever a estrutura de qualquer proteína apenas a partir de sua sequência de aminoácidos”. Agora, após cinco décadas, cientistas conseguiram criar um software impulsionado por inteligência artificial que consegue concretizar o sonho de Anfinsen – compor estruturas proteicas precisas com base em informações de seus aminoácidos inseridas no programa. A descoberta tem implicações tão grandes que recebeu o destaque da revista Science – descoberta do ano de 2021.
As proteínas são os burros de carga da biologia. Elas contraem nossos músculos, convertem alimentos em energia celular, transportam oxigênio em nosso sangue e lutam contra invasores em nossos organismos. No entanto, apesar de seus talentos variados, todas as proteínas são compostas pela mesma forma básica: uma cadeia linear de até 20 tipos diferentes de aminoácidos, unidos em uma sequência codificada em nosso DNA. Depois de ser montada em “fábricas” chamadas ribossomos, dentro de nossas células, cada corrente se dobra em uma forma 3D única e extremamente complexa. São essas formas que determinam como as proteínas interagem com outras moléculas e definem seus papéis na célula.
Entretanto, as estruturas das proteínas só podiam ser determinadas por meio de meticulosas análises de laboratório. Agora, pesquisas que unem biologia molecular à engenharia de software conseguiram calcular rapidamente a forma de dezenas de milhares de proteínas com base na sequência de seus blocos básicos. Nos anos 1970, o trabalho de Anfinsen sugeriu que as formas finais das proteínas eram determinadas pelas interações entre seus aminoácidos. Mas cientistas daquela década nunca conseguiram comprovar a hipótese devido ao número de possíveis interações entre cada elo da cadeia – a hemoglobina em nosso sangue, por exemplo, é formada por uma cadeia de 574 aminoácidos; se cada aminoácido interage com todos os demais, imagine o número de interações possíveis entre todos esses blocos.
Mas, na natureza, cada proteína se dobra em apenas uma forma distinta, geralmente em um piscar de olhos. Os primeiros modelos de computador para prever como uma determinada proteína se dobraria foram desenvolvidos na década de 1970, mas conseguiam prever apenas a forma que seria assumida por segmentos pequenos de aminoácidos. Em 1994, os esforços para programar modelos de computador se tornaram uma espécie de competição bienal no evento Avaliação Crítica da Predição da Estrutura de Proteínas (CASP – Critical Assessment of protein Structure Prediction).
O desempenho foi melhorando lentamente, em uma progressão linear, até que, em 2018, o software baseado em inteligência artificial chamado AlphaFold entrou em cena. O programa desenvolvido pela empresa britânica DeepMind, de propriedade do Google, utiliza como bancos de dados as estruturas 3D montadas ao longo das décadas. Com base nesse acervo, o software “treinou” a si mesmo para reconhecer a forma de interação dos aminoácidos.
Agora, o AlphaFold se tornou tão competente em prever o formato de proteínas quanto as técnicas “manuais” utilizadas pelos cientistas (cristalografia de raios-x, espectroscopia de ressonância magnética nuclear e microscopia crioeletrônica) – mas consegue fazer isso milhares de vezes mais rápido. Este ano surgiram outros softwares de previsão do formato de proteínas com base em inteligência artificial, como o RoseTTAFold. Já foram mapeadas mais de 350 mil proteínas encontradas no corpo humano (44% do total da variedade proteínas que possuímos).
